Homology Groups in Markush Structures
Homology groups represent sets of homologous substructures in Markush structutes (e.g., alkyl, aryl, heterocycle, etc.). Read the user's guide about homology groups and editing their properties in MarvinSketch and in Marvin JS.
Currently, JChem search supports homology groups on the query and target side, but not on both sides at the same time. Various restrictive properties can also be specified for homology groups.
Contents
Definition of homology groups
Homology groups are represented by Pseudo atoms, labeled with common chemical annotations of these groups. Some groups have multiple alias names (abbreviations, alternative spellings). The names are case insensitive, spaces might be inserted.
There are two major types of homology groups regarding the way of their definition:
-
Built-in homology groups are defined by specific structural properties of the group. These groups are not enumerated during the search, but appropriate substructures are recognized as fulfilling the requirements for such a structure. The possible number of covered structures is usually infinite, unless the number of atoms is limited. Examples of built-in groups are alkyl, aryl, heterocycle, etc.
-
User-defined homology groups are explicitly defined, and only the listed substructures can match these homology groups. The definition is given in the form of an R-group definition, in which any generic Markush feature can be used. There are some Predefined groups, and new 'User-defined' groups can also be added. These 'User-defined' definitions can be customized by the user, and they can be context-specific. (E.g. Protecting group definitiondepends on which functional group it protects.)
1. Built-in homology groups
Table 1. shows the properties of the built-in homology groups. Each group describes a set of substructures having specific features. These features are shown in the table as "compulsory" parts. Some groups also allow optional parts that might be present in the substructure that matches the homology group.
Table 1. Built-in homology groups
| Group name (alias names) | Description | Example | Note |
|---|---|---|---|
| Alkyl (CHK) | - only carbon and hydrogen atoms - at least one carbon atom - only single bonds - no ring bonds - optional: connection points at arbitrary positions |
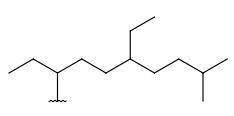 |
|
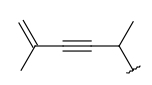
| Alkenyl (CHE) | - at least one double bond, no triple bonds - at least two carbon atoms - otherwise same as for Alkyl |
 |
|
| Alkynyl (CHY) | - at least one triple bond - at least two carbon atoms - optional: double bonds - otherwise same as for Alkyl |
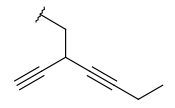 |
|
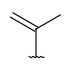
| CarbonChain (AcyclicCarbon, CarbonTree) | - any connected acyclic hydrocarbon (branched or unbranched) - optional: connection points at arbitrary positions |
 |
renamed since version 22.18.0 (was CarbonTree) |
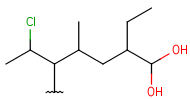
| HeteroSubstitutedAlkyl (HSA) | - at least one heteroatom - at least one carbon atom - only single bonds - no ring bonds- each heteroatom is connected to a single carbon atom and (optionally) hydrogens - optional: connection points at arbitrary carbon atoms |
 |
|
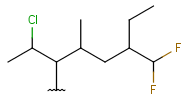
| Haloalkyl | - each heteroatom is halogen - otherwise same as for HeteroSubstitutedAlkyl |
 |
|
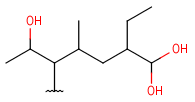
| Hydroxyalkyl | - each heteroatom is oxygen - otherwise same as for HeteroSubstitutedAlkyl |
 |
|
| Cyclyl (AnyCyclyl, AnyRing) | - monocyclic or fused ring(s) without any restrictions - optional: connection points at arbitrary positions |
 |
|
| Aryl | - monocyclic or fused ring(s) - at least one ring should be aromatic - cannot have external connection on an aliphatic ring - optional: double or triple bonds in aliphatic rings - optional: arbitrary number of connection points, but all must be on aromatic rings |
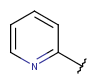 |
since version 17.21.0 |
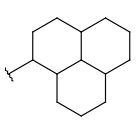
| Carbocyclyl | - only carbon and hydrogen atoms - otherwise same as for Cyclyl |
 |
since version 20.20.0 |
| Carboaryl (ARY) | - only carbon and hydrogen atoms - otherwise same as for Aryl |
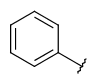 |
|
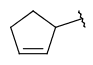
| Carboalicyclyl (CYC, Cycloalkyl) | - monocyclic or fused aliphatic ring(s) - only carbon and hydrogen atoms - optional: double or triple bonds - optional: connection points at arbitrary positions |
 |
|
| Heterocyclyl (Heterocycle) | - at least one heteroatom - at least one carbon atom - otherwise same as for Cyclyl |
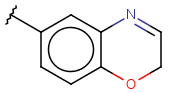 |
since version 15.7.6 |
| Heteromonocyclyl | - monocyclic ring - otherwise same as for Heterocyclyl |
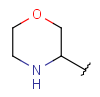 |
since version 17.21.0 |
| Fusedheterocyclyl (HEF, Heteropolycyclyl, FusedHetero) | - fused rings - otherwise same as for Heterocyclyl |
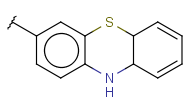 |
renamed since version 22.18.0 (was FusedHetero) |
| Heteroaryl | - at least one heteroatom - at least one carbon atom - at least one heteroatom in aromatic ring - otherwise same as for Aryl |
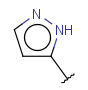 |
since version 17.21.0 |
| Heteromonoaryl (HEA) | - monocyclic ring - otherwise same as for Heteroaryl |
|
|
| Fusedheteroaryl (Heteropolyaryl) | - fused rings - otherwise same as for Heteroaryl |
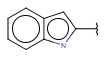 |
since version 17.21.0 |
| Heteroalicyclyl | - monocyclic or fused aliphatic ring(s) - at least one heteroatom - at least one carbon atom - optional: double or triple bonds - optional: connection points at arbitrary positions |
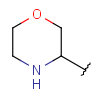 |
since version 17.21.0 |
| Heteromonoalicyclyl (HET) | - monocyclic ring - otherwise same as for Heteroalicyclyl |
|
|
| Fusedheteroalicyclyl (Heteropolyalicyclyl) | - fused rings - otherwise same as for Heteroalicyclyl |
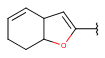 |
since version 17.21.0 |
| RingSegment | - part of a ring where every atom has only two ring bonds - not a whole ring - optional: non-ring connections (substituents) |
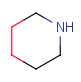 |
|
| Halogen (HAL) | - a single halogen atom | F, Cl, Br, I | |
| Metal (MX) | - any metal atom | U, K, Fe, Na, Ni, Al, ... | |
| AlkaliMetal (AMX) | - alkali or alkaline earth metal atom | Na, K, Ca, Mg, ... | |
| TransitionMetal (TRM) | - transition metal atom excluding lanthanum | Fe, Ni, Zn, Co, Hg, W, ... | |
| Lanthanide (LAN) | - lanthanide atom (including lanthanum) | Nd, Ce, Pr, ... | |
| Actinide (ACT) | - actinide atom (including actinium) | U, Th, Pa, ... | |
| OtherMetal (A35) | - group IIIa-Va metal atom | Al, Ga, ... | |
| AnyAtom | - a single atom except for hydrogen | C, N, O, P, S, ... | |
| AnyGroup (XX) UnknownGroup (UNK) |
- any structure (excluding a single hydrogen atom) | AnyGroup and UnknownGroup are equivalent since version 23.16.0 |
Subset rules between homology groups
- Alkyl, Alkenyl, Alkynyl are subsets of CarbonChain
- all cyclic groups are subsets of Cyclyl
- all carbocyclic groups are subsets of Carbocyclyl
- all heterocyclic groups are subsets of Heterocyclyl
- Haloalkyl and Hydroxyalkyl are subsets of HeteroSubstitutedAlkyl
- AlkaliMetal, TransitionMetal, Lanthanide, Actinide, OtherMetal are subsets of Metal
- all homology groups are subsets of AnyGroup / UnknownGroup
2. User-defined homology groups
Besides the built-in homology groups, users can also define custom groups. User-defined homology groups are represented by R-group definitions, and during search the pseudo atoms of user-defined homology groups are translated to the corresponding R-group definitions.
These group definitions are customizable, the user can modify them or can make new definitions as well. Group names are treated as case insensitive, but in case sensitive file systems the definition files should be lowercase.
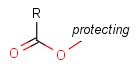
Protecting group
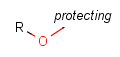
There is a special, predefined (user-defined) homology group that is readily available. It is called Protecting or PRT.
Protecting groups' definition file contains several definitions, each for protecting different functional groups. The protected functional group is defined by the neighborhood of the R-atom. When the R-atom has the same neighborhood as the "protecting" pseudo atom, then the group is replaced by the R-atom.
The conversion processes the group definitions in their order in the file. This means that more specific environments should be placed earlier. For example, a carboxyl protecting group definition should precede an alcohol definition, otherwise the alcohol definitions will be applied instead. Currently, they are located in the following order:
-
amino
-
carboxyl
-
alcohol
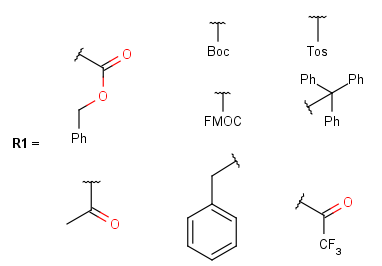
The system cannot handle protecting groups having more than one attachment point, or groups where the heavy atoms of the functional group should be changed by the substitution. The readily available definitions contain amine, carboxyl and hydroxyl protecting groups.
Some examples with different functional groups protected can be found in Table 2.
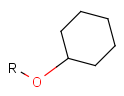
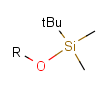
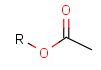
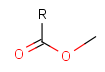
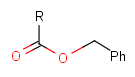
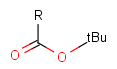
Table 2. Protecting group examples
| Protecting group | Represented examples | ||
|---|---|---|---|
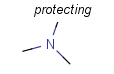 |
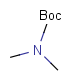 |
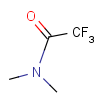 |
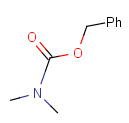 |
 |
 |
 |
 |
 |
 |
 |
 |
Markush enumeration
To enable the enumeration of homology groups, the "Homology Enumeration" option of Markush enumeration has to be switched on. Otherwise, the homology groups are kept as pseudo atoms. This latter option might be useful for showing that these structures cannot be fully enumerated.
Built-in homology groups
For the built-in homology groups, a small set of example structures are used in the case of enumeration. These examples are characteristic to the homology group and encompass simple and large structures as well. They are provided as an R-group definition, similarly to the definition of user-defined homology groups.
We have to emphasize that these example structures are used only for enumeration and do not affect searching. As noted earlier, arbitrary structures fulfilling the requirements for the homology group will match such a target.
Enumeration definitions contain two attachment points by default. After enumeration these are the atoms which connect to the first two neighbors of the group. If the enumerated homology group's pseudo atom has more than two connections, then further attachment points are added. These are put on atoms that have free valence and comply the requirements for externally connecting atoms of the given group. E.g. for Aryl, only aromatic ring atoms can be the connection points. The atoms of the definition are investigated in the order of the atom numbers. If a definition does not have the sufficient number of such atoms, then it is rejected. When every definition of the homology group is rejected, an exception is thrown showing that the given homology group does not have any valid enumeration definition.
User-defined homology groups
Enumeration of user-defined homology groups uses the same customizable R-group definitions as searching. User-defined homology groups should have the same number of connections as in the definitions.
Properties of homology groups
Some homology groups can have important properties. You might want to specify if the alkyl chain is branched, or any deuterium atoms are present. The homology groups have a special property editing dialog where you can set the different properties. They include the followings (with the group to which it may be applied):
-
Deuterium and tritium count: for all homology groups. The value should be given as e.g. D1-4T3, meaning the group contains up to 4 deuterium atoms and 3 tritium atoms.
-
Text notes: for all homology groups (see details in next section).
-
Branching: for chain homology groups (BRA for branched, STR for straight chain).
-
Size: for chains. Chains are marked as low (C1-6. LO), mid (C7-10, MID) or high (C11-, HI) according to the length of the chain.
-
Saturation: for ring groups. They can be marked as saturated or unsaturated.
-
Ring type: for ring groups. They are marked as monocyclic (MON) or multicyclic (FU), or can be marked as 'not specified'.
Not specifying a property means that there is no restriction on that property.
Table 5. Available properties of homology groups.
| Category | Homology groups | Size | Branching | D/T count | Ring type | Saturation | Additional Text Notes |
|---|---|---|---|---|---|---|---|
| Acyclic groups | Alkyl, Alkenyl, Alkynyl, CarbonChain |  |
|
|
 |
|
|
| HeteroSubstitutedAlkyl, Haloalkyl, Hydroxyalkyl | |
|
|
|
|
|
|
| Cyclic groups | Aryl, Carboaryl | |
|
|
|
|
|
| Carboalicyclyl | |
|
|
|
|
|
|
| Heteroaryl | |
|
|
|
|
|
|
| Heteromonoaryl, Fusedheteroaryl | |
|
|
|
|
|
|
| Heteroalicyclyl | |
|
|
|
|
|
|
| Heteromonoalicyclyl, Fusedheteroalicyclyl | |
|
|
|
|
|
|
| Heteromonocyclyl, Fusedheterocyclyl | |
|
|
|
|
|
|
| Cyclyl, Carbocyclyl, Heterocyclyl | |
|
|
|
|
|
|
| RingSegment | |
|
|
|
|
|
|
| Atomic groups | Halogen, Metal, AlkaliMetal, TransitionMetal, Lantanide, Actinide, OtherMetal, AnyAtom | |
|
|
|
|
|
| Special groups | AnyGroup, UnknownGroup | |
|
|
|
|
|
Additional text notes
Text format: letters denoting different parameters followed by number ranges. These entries are separated by commas. Specification of attachment atom type is also possible.
| Parameter | Description |
|---|---|
| Z | Number of single bonds (from version 22.20.0) |
| E | Number of double bonds |
| Y | Number of triple bonds |
| A | Number of aromatic bonds (from version 22.20.0) |
| C | Number of carbon atoms |
| Heteroatom symbol | Number of heteroatoms of the specified element (e.g., N1-3) |
| X | Number of heteroatoms not defined otherwise |
| Q | Number of heteroatoms, also including the ones defined otherwise (from version 20.19.0) |
| HAL | Number of occurrences of halogen atoms (e.g., HAL1-5) |
| NR | Number of rings (according to SSSR) |
| RA | Number of ring atoms |
| >Atomic symbol | One attachment to an atom of the specified element |
| >>Atomic symbol | Multiple attachments to atoms of the specified element |
| FRC | Number of fused ring connections in the ring system matched by the homology group (from version 24.3.0) |
| BRC | Number of bridge ring connections. Rings connection counts are calculated based on SSSR (smallest set of smallest rings), so a bridge will count as 1 connection instead of 3 (from version 24.3.0) |
| SRC | Number of spiro ring connections (from version 24.3.0) |
Example: N1-3,NR4,E1-2,>>C
Customizing homology groups
Location of user-defined homology group definition files
The default location of chemaxon_home directory of the user on different platforms:
-
Windows: %USERPROFILE%\chemaxon\ (in other words ..\Users\<USERNAME>\chemaxon)
-
Unix/Linux: ~/.chemaxon/
Location of "User-defined" (for search and enumeration) user-defined homology group definition files: chemaxon_home/homology/user_def_groups/
Location of "Enumeration-only" user-defined homology group definition files: chemaxon_home/homology/enumeration_only/
Note: Create the above two directories if they do not exist.
In order to define a new user-defined homology group, you should add its definition as an R-group to the directory chemaxon/enumeration/homology/user_def_groups within the JAR file using a new name that does conflict with existing groups. These groups are represented by these definitions during search and enumeration as well.
In order to customize the enumeration of existing homology groups, you should change the corresponding file in the directory chemaxon/enumeration/homology/enumeration_only within the JAR file
Defining new user-defined homology groups
1. Draw the desired group definition in MarvinSketch and save as mrv; the name of the new group should be specified by the name of the file; the name of the file must be in lower case;
See example nucleobase.mrv below:
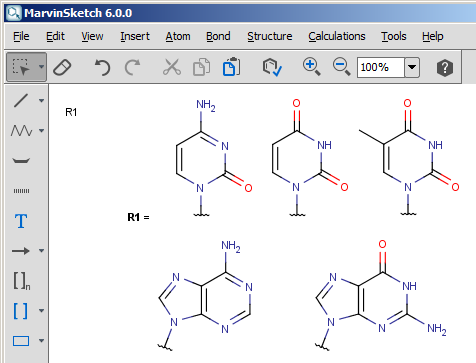
1. copy the mrv file into chemaxon_home/homology/user_def_groups/ .
The files of enumeration-only type User-defined groups should be placed into the directory chemaxon_home/homology/enumeration_only/ .
Modifying the predefined homology groups
Modifying these files will affect searching/enumeration in the case of predefined (user-defined) groups and only the enumeration in the case of built-in groups.
The modified definition or the newly added group can also be dependent on the neighborhood (context-sensitive) as in the case of Protecting groups.
The modification of these definitions can be executed:
-
the same way as described above for the creation of the NEW User-defined homology (or protecting) groups, but the name of the mrv file must be the same as the built-in file name within com.chemaxon-enumeration.jar; copy the mrv file into chemaxon_home/homology/user_def_groups/
-
or by modifying the existing default file from com.chemaxon-enumeration.jar
1. Copy protecting group definition to the user's chemaxon library: e.g. from .../com.chemaxon-enumeration.jar/chemaxon/enumeration/homology/user_def_groups/protecting.mrv to chemaxon_home/homology/user_def_groups/
1. Open the newly copied file in the user's directory with MarvinSketch.
1. A dialog appears asking the index of molecule to open. Enter 1 because this contains the amino protecting group definition. If the proper molecule number is not known, all the definitions can be displayed using MarvinView.
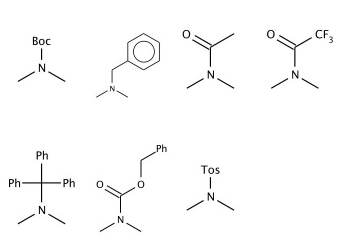
1. Overwrite the structures, e.g. delete the FMOC group, see Table 4. The new definition will be used in searching and enumeration, see Table 4.
The files of enumeration-only type user-defined groups must be placed into the directory chemaxon_home /homology/enumeration_only/ .
If you would like to have different definitions for searching and enumeration of a user-defined group, then a separate file should be specified under the same file name in the " enumeration_only " dictionary as well. In this case the content of the " user_def_groups " will be used during searching and the content of the " enumeration_only " for enumeration.
If a definition is modified it comes into effect immediately, however the addition of a new group requires a restart of the Java Virtual Machine.
Table 4. Modifying amino protecting group definitions.
| New definition | Sample Markush file | Enumerated structures |
|---|---|---|
 |
 |
 |