Reactor FAQ
Here we collected Frequently Asked Questions about Reactor for you.
-
I am familiar with SMILES and SMARTS, does Reactor support them?
-
I do not like SMILES and SMARTS, can I use Molfile, SDfiles and Rxnfiles with Reactor?
-
Should I mark the substitutable parts with R-atoms in the reaction scheme?
-
Is the order of the reactant files important in a multimolecular reaction?
-
Is it possible to build the functionality of Reactor into my application?
-
Is there a simple desktop GUI for end user chemists for hardcore virtual synthesis?
-
I found, that one of your reactions generates unfeasible products, what should I do?
-
How can I specify, that a functional group in a reaction is an amine but not amide?
-
What is the difference between reactions in Reactor and transforms in Standardizer?
What is Reactor?
Reactor is virtual synthesis tool transfoming molecules to products according to given reaction schemes. It is part of Chemaxon's JChem toolkit.
What is Reactor good for?
You can discover the virtual compound space, enumerate combichem libraries, predict metabolites, or create other molecule transformation-based applications with Reactor.
Will Reactor run on my system?
Since Reactor is written in pure Java, it can be used on any platforms supported by the Java Runtime Environment version 1.6 or later, including most Windows, Mac, Linux and Unix platforms.
I am familiar with SMILES and SMARTS, does Reactor support them?
Yes, you can even run a reaction from the command line.
I do not like SMILES and SMARTS, can I use Molfile, SDfiles and Rxnfiles with Reactor?
Yes, Reactor supports lots of formats, the choice is yours.
What are map numbers?
Map numbers assign the corresponding atoms on the two sides of the reaction arrow of the scheme.
What are orphan atoms?
Orphan atoms appear only on one side of the reaction arrow. They appear or disappear during the reaction.
Which atoms should I map?
By default, Reactor expects, that all atoms having changing bonds are mapped. Orphan atoms must be mapped, since they have changing bonds. However, Reactor can handle other mapping styles too (when orphan atoms must not be mapped), and provides a built in automapper.
Can my reactions contain R-groups?
Yes, the reaction scheme can contain R-group definitions (appearing on both sides of the reaction arrow). Use R-groups as you use them in R-group queries.
Should I mark the substitutable parts with R-atoms in the reaction scheme?
No, Reactor uses the reactants of the scheme as queries to find reaction sites in the input molecules. See the the Query Guide for more details on queries.
What file formats are supported by Reactor?
Reaction definitions can be written in MRV, RXN, RDF or SMIRKS/SMARTS formats, the molecules in MRV, MOL, SDF, CML or SMILES.
Is there a limit for number of reactants?
No. Reactor supports monomolecular, bimolecular, trimolecular and even higher order reactions, no limit.
Is the order of the reactant files important in a multimolecular reaction?
Yes. Reactor follows the visual order of the reactants in the reaction scheme (left to right, top to down). Thus, the leftmost reactant is the first one. Be sure to specify the input files in that order.
Can I run Reactor in batch mode?
Absolutely, you can generate large libraries in a single step.
What about the speed of the library generation?
Reactor, in general, is lightning fast. Although, the exact speed depends on the reaction schema and the calculations defined in the rules, the slowest step is usually writing the results into a hard disk. In case of SMILES output - which is a relatively small molecule description format - the speed can be higher than half million reactions/hour even on an ordinary notebook. In case of a database output, the speed is usually a bit less but it is still possible to generate some millions of compounds overnight.
Is it possible to build the functionality of Reactor into my application?
Yes, Reactor is delivered with full API, you can equip your own applications with virtual synthesis features.
Can I run reactions from a Unix shell?
Yes, there is a script and a batch file as well for those who prefer command line tools, just type react .
Is there a simple desktop GUI for end user chemists for hardcore virtual synthesis?
Will be available soon, will are working on it.
How can I evaluate Reactor?
Download Reactor from our website.
Do you have some batch mode examples to start with?
Yes, try our working examples.
What are sequential and combinatorial modes?
Let's take a bimolecular reaction example, where the reagents are located in two SDfiles. In combinatorial mode, all molecules of the first file are reacted with all molecules of the second file. In sequental mode, the two first molecules are reacted, then second ones, and so on.
What are those "smart" reactions?
A "smart" reaction contains the corresponding "synthetic knowledge" in rules. These rules provide the basis of chemically feasible virtual synthesis.
In what format are the rules written?
The rules are written in Chemical Terms, a simple language containing logic, arithmetic and lots of chemistry related functions to create complex expressions.
What is the Reactivity rule?
The functional groups of the generic reaction scheme and their required properties can be refined in the Reactivity rule to correctly specify the reactive sites.
What is the Selectivity rule?
The Selectivity rule helps to determine the main product when more than one reactive site is found on a reactant. The region with the highest Selectivity value is considered to provide the main product. Regioselectivities like ortho/para or meta directing are usually defined in the Selectivity rule.
What is the Tolerance value?
The Tolerance value makes it possible to have more than one main product in certain cases. Take an aromatic electrophilic substitution reaction as an example, in which the electrophilic localization energy on the aromatic ring is the lowest in the para position, but it is almost as negative in the ortho position. If the difference is less than the tolerance, both ortho and para isomers are generated as main products.
What is the Exclude rule?
Use the Exclude rule to skip reactants containing functional groups causing side reactions. For example, you can avoid generating products from molecules having alcohol or amine functionalities, if you exclude starting compounds having nucleophilic groups by this rule.
Can I use Reactor with schemes without any rules defined?
Can I skip the rules of a reaction?
Yes, you can skip the rules to examine their effects.
Can I change the rules?
Yes. The rules can be modified to improve the quality of the virtual reactions.
Can I generate drug-like compounds only?
Drug-likeness filters can easily be written in Chemical Terms. Although you could, you should not add a drug-likeness filter (or similar) to the rules. Specific synthesis goals should not appear in the reactions. You can post-filter the products or use Synthesizer (part of Reactor Pro) to specify custom conditions for each reaction step. This will not hurt the integrity and reusability of the reactions.
Do you provide a ready-to-use reaction library?
Chemaxon provides a library of several important preparative reactions for the Reactor users. Download it from the Reactor web-page.
I found, that one of your reactions generates unfeasible products, what should I do?
Please inform us about the problem to be able to provide better virtual reactions for the community. Check our contact details here.
How can I specify, that a functional group in a reaction is an amine but not amide?
There are various ways to define that, let's see them by an acylation reaction example:
- Write the reaction scheme in recursive SMARTS.
[H:4][N;X3;!$(NC=O):3].O=[C:1][Cl:2]>>O=[C:1][#7:3].[Cl:2][H:4]
- Define the reaction scheme with an R-group and set its occurence to 0.
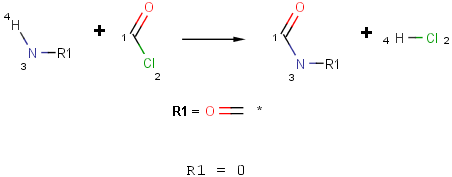
- Refine the functional group definition in the Reactivity field using the match function of Chemical Terms.
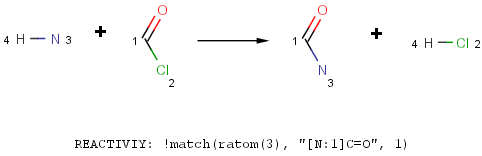
What is the difference between reactions in Reactor and transforms in Standardizer?
Standardizer is for the canonicalization and beautification of molecules, Reactor is for feasible virtual synthesis.
Standardizer supports transforms:
-
single input molecules
-
one input molecule is converted to one output molecule
-
all corresponding groups of an input molecule are transformed at once
-
no advanced synthetic feasibility prediciton
Reactor supports virtual reactions:
-
both unimolecular and multimolecular reactions
-
one reaction can produce multiple products (including isomers)
-
reacts one reaction site at once (since it affects the reactivity/selectivity features for the next step)
-
can generate synthetically feasible products (supports reactivity, selectivity and exclude rules)
Can't find an answer to your question? Visit our Reactor Getting Help and Support page and check how to contact us.