Jarvis-Patrick clustering
This manual gives you walk-through on the Jarvis-Patrick clustering algorithm:
Introduction
Jarp performs variable-length Jarvis-Patrick clustering based on fingerprints and/or other data stored in a database table or a file. The software can also be used for calculating diversity measures, like average and minimum dissimilarity of a compound library. This document mentions molecules as the entities to be clustered, but the software can also be used for other types of objects.
The original Jarvis-Patrick algorithmproceeds as follows:
-
Calculate the set of K nearest neighbors for each structure. Note that the structure is set as its 0th neighbor.
-
Two structures cluster together if
-
they are in each others list of nearest neighbors
-
they have at least Kmin of their K nearest neighbors in common.
-
Kmin and K are adjustable parameters. Several variations of the Jarvis-Patrick algorithm exist. The one applied in Jarp is based on variable-length lists of nearest neighbors:
-
For each structure, collect the set of nearest neighbors that has a dissimilarity (distance) less than a T threshold value.
-
Two structures cluster together if
-
they are in each others list of nearest neighbors
-
they have at least Rmin of their nearest neighbors in common, where Rmin is a ratio of the length of the shorter list
-
T and Rmin are adjustable parameters. The advantages of the latter method are explained by Brown and Martin and Barnard. The original algorithm has the following shortcomings:
-
Nearest neighbors of equal similarity to others already in the list maybe excluded because the list already has K elements. This may subdivide large clusters consisting similar molecules.
-
It has the tendency to produce too many singletons, because in many cases similar structures have less than Kmin nearest neighbors in common.
-
Molecules in sparsely populated areas tend to cluster together.
The application of variable-length lists solve the above problems.
Nearest neighbor searching finds molecules that are similar to the query object. The calculation applies the Tanimoto (or Jaccard) coefficient that is calculated by the following formula in the case of binary fingerprint (bit string) input:
T(A,B) = NA&B/(NA+NB-NA&B) where NA and NB are the number of bits set in the bit strings of molecules A and B, respectively, NA&B is the number of bits that are set in both.
When only binary fingerprints are used for the calculation of the dissimilarity between molecules, then the formula of the dissimilarity of molecule A and B is
D(A,B) = 1-T(A,B) where T(A,B) is the Tanimoto coefficient for molecule A and B.
When other types of columns are (also) used, a weighted Euclidean distance calculation is applied:
D(A,B) = sqrt{[1-T(A,B)] + w1[C1(A)-C1(B)]2 + w2[C2(A)-C2(B)]2 + ...} where
-
w1, w2, ... are weights
-
T(A,B) is the Tanimoto coefficient for molecule A and B
-
Ci(A) is the value of descriptor i of molecule A.
-
sqrt is the square root function.
Instead of the brute force method, Jarp applies heuristics to avoid calculating all pairwise dissimilarity calculations and neighbor list comparisons. According to our measurements, the speed of clustering is O(n1.5).
Usage
You can use Jarvis-Patrick clustering via the jarp command line tool as follows:
jarp [<options>]Prepare the usage of the jarp script or batch file as described in Preparing the Usage of JChem Batch Files and Shell Scripts.
You can also call the JarvisPatrick Java class directly as follows:
-
Under Win32 / Java 2 (assuming that JChem is installed in c:\jchem):
java -cp "c:\jchem\lib\jchem.jar;%CLASSPATH%" chemaxon.clustering.JarvisPatrick [<options>] -
Under Unix / Java 2 (assuming that JChem is installed in /usr/local/jchem):
java -cp "/usr/local/jchem/lib/jchem.jar:$CLASSPATH" \ chemaxon.clustering.JarvisPatrick [<options>]
Because the utility has many parameters, it may be reasonable to create a shell script or a batch file for calling the software.
Options
The following options can be applied for clustering:
General options: -h --help this help message -d --driver <JDBC driver> JDBC driver -u --dburl <url> URL of database -l --login <login> login name -p --password <password> password -P --proptable <tablename> name of property table -s --saveconf save settings into ~/.jchem Input options (default: standard input): -i --input <filepath> input file to cluster (text file input) -q --query <sql> SQL query for reading input (database input) Output options (default: standard output): -o --output <filepath> output file path (text file output) -a --statement <sql> SQL statement for inserting results (database output) -x --central calculate and sign central objects -y --singlet singletons get negative cluster ids -z --statistics print statistics -Z --only-statistics print only statistics -v --verbose verbose output Data properties -m --dimensions <dim> number of floating-point descriptors -f --fingerprint-size <bits> binary fingerprint size in bits. fpsize should be a multiple of 32-w --weights <w1> <w2> ... the weights of the floating-point descriptors -g --generate-id generate id for each compound. Clustering conditions -t --threshold <threshold> maximum dissimilarity of two compounds -c --common <ratio> minimum ratio of common neighbors of two compounds. {warning} Without a valid license key, the software is in demo mode and maximum 1000 structures can be retrieved from the database.
Input options
The software may import data from either a text file ( --input ) or a database ( --query ). The input data must contain the following columns:
| Columns | Type | Content |
|---|---|---|
| Id | Integer numbers | Id of compounds (Optional in text files) |
| fp1, fp2, fp3 ... | Integer numbers | Binary fingerprints in integer number blocks The number of fp. columns is fp. length / 32 (Optional) |
| d1, d2, d3, ... | Floating point numbers | Other descriptors (Optional) |
Comments:
-
Pharmacophore fingerprints can be generated using the GenerateMD tool. These fingerprints are not binary, so they have to be specified as other descriptors. See an example for combining GenrateMD and Jarp.
-
At least one binary fingerprint column or descriptor column is required.
-
Use the --generate-id option if the id column is missing from the input data.
-
Text input files can be created using the GenerateMD application. For example:
generatemd c structures.smi -k CF -c cfp.xml -D -o fingerprints.txt -
In the case of text input, the delimiter between two numbers should be space or tab (comma is not allowed).
-
The cd_id and cd_fpi columns in JChem's structure tables are appropriate as input.
-
In the case of database input, an SQL select statement is needed to retrieve the columns. For example:
jarp -q "SELECT cd_id, cd_fp1, cd_fp2, cd_fp3, cd_fp4, cd_fp5, cd_fp6 FROM structures" ...For the sake of readability only 6 fp. columns is applied in the above example, but usually this number is much higher. You may also modify the order of the results by appending " ORDER BY CD_ID" to the SQL query.
-
It is important to place the query statement between quotes because it contains spaces.
Output options
The software can write the results of clustering into either a text file ( --output ) or a database table ( --statement ). The exported data contains the following columns:
| Columns | Type | Content |
|---|---|---|
| Id | Integer numbers | Identifier of compounds |
| Clid | Integer numbers | Cluster identifier |
| Centr | Integer numbers | Displays whether the object is central |
The last column is written only if the --central option is specified. A central object has the smallest sum of dissimilarities to the other objects in the cluster. Central object calculation slows down the application significantly.
Comments for text output:
-
The Id and Clid columns are the same as in the case of database output.
-
A "@" symbol is used to designate the central objects of the clusters
Comments for database output:
-
A precondition of database output is the existence of a database table that contains the above columns. Create the database table before starting the calculation. Examples for table creation:
-
If the result will not contain central objects:
CREATE TABLE clusters ( cd_id INTEGER NOT NULL PRIMARY KEY, cluster_id INTEGER) -
If the result will contain central objects:
CREATE TABLE clusters ( cd_id INTEGER NOT NULL PRIMARY KEY, cluster_id INTEGER, central SMALLINT)
-
-
Before clustering, make sure that the table is empty. The SQL DELETE statement may be applied for deleting the rows in a database table.
Example for deleting all rows:
DELETE FROM clusters; -
In the case of database output, an SQL statement is needed to be specified for Jarp ( -a option), which inserts the rows containing the results. For example:
jarp -a "INSERT INTO clusters(cd_id, cluster_id, central) VALUES(?,?,?)" ...The ? symbols will be substituted with the corresponding values.
-
If the table is filled with the results, the clusters may be retrieved using SQL SELECT statements. For example:
SELECT * FROM clusters WHERE cluster_id = 1 -
It is important to place the import statement between quotes because it contains spaces.
-
The central column is 1 if the object is central, 0 otherwise.
Parameters
The following parameters can be set for Jarvis-Patrick clustering:
--fingerprint-size The number of binary fingerprint columns multiplied by 32 (because the bit-length of integer numbers is 32 in Java)
--dimensions Specifies the number of other columns. If only binary fingerprints are used in the clustering process, then this parameter doesn't have to be set.
--weights When other columns are used, a weighted Euclidean distance calculation may be applied. If there are also binary fingerprint columns, weights are relative to the Tanimoto coefficient calculated from the binary fingerprints (the Tanimoto coefficient has a weight of 1.0).
--threshold Same as T in the Introduction. The higher the threshold the longer the lists of neighbors that has to be stored in memory for each compound during clustering. Too long lists of neighbors can cause memory problems.
--common Same as Rmin in the Introduction.
Loading and saving settings
By default, the heap size in some Java runtime environments is limited to 64MB, so you may run out of memory easily.It would be inconvenient to enter all of the parameters of the jarp script at each run. To overcome this problem, it is possible to save some of the settings that are not changed frequently in the .jchem file stored in the user's home directory. Use the --saveconf option to store the following settings:
-
JDBC driver's class name ( --driver )
-
JDBC URL of database ( --dburl )
-
Login name ( --login )
-
Password ( --password )
-
Binary fingerprint size ( --fingerprint-size )
JChemManager saves the modified settings that needs for the database connection. If you successfully entered into the database using JChemManager, then you don't need to set connection for Jarp manually.
Clustering statistics
Optionally Jarp can print clustering statistics into the standard output or the given output file. The parameters that enable statistics printing are --statistics or --only-statistics . (The latter one doesn't allow to print information on individual compounds.)
The following data will be printed:
-
Number of objects
-
Number of clusters
-
List of clusters (cluster id, size, central object)
-
Statistics on pairwise dissimilarity values:
-
average
-
minimum
-
maximum
-
The calculation is significantly slower if statistics is enabled, since all pairwise dissimilarity values have to be calculated. (Heuristics cannot be applied.)
Running RNN search and clustering separately
Setting the minimum proportion of common neighbors ( --common option) correctly, is important in fine tuning the clustering process. Since nearest neighbor searching is much more time consuming than the clustering stage, it is reasonable to separate the two processes. In that case clustering can be run several times with different --common settings.
nneib is a command line application that collects and stores the nearest neighbor list in a text file. If this file is fed into Jarp, the nearest neighbor search is omitted. NNeib has the same command line parameters than Jarp, however, the --common , --statistics , --only-statistics , --singlet options are not available.
Run
nneib -hfor help on input parameters.
If the threshold value ( --threshold ) is not specified for Jarp, then
-
it expects a NNeib generated neighbor list in the input text file
-
in the case of the --statistics and --only-statistics options, diversity statistics is not printed
-
central object calculation ( --central ) is not available
-
the following parameters have to be specified only for NNeib:
--query
--weights
--generate-id
--dimensions
Examples
In the examples it is supposed that all connection parameters are set and stored by JChemManager (or a previous saving by Jarp):
-
A batch file (Windows) for reading from a database and writing to the standard output:
set QUERY="SELECT cd_id, cd_fp1, cd_fp2, cd_fp3, cd_fp4, cd_fp5, cd_fp6, cd_fp7, cd_fp8, cd_fp9, cd_fp10, cd_fp11, cd_fp12, cd_fp13, cd_fp14, cd_fp15, cd_fp16 FROM structures WHERE cd_id < 10000" jarp -q %QUERY% -t 0.1 -c 0.3 -f 512 -
A UNIX shell script for reading from a database and writing to another table:
QUERY="SELECT cd_id, cd_fp1, cd_fp2, cd_fp3, cd_fp4, cd_fp5, cd_fp6, cd_fp7, cd_fp8, cd_fp9, cd_fp10, cd_fp11, cd_fp12, cd_fp13, cd_fp14, cd_fp15, cd_fp16 FROM structures WHERE cd_id < 10000" INSERT="INSERT INTO clusters(cd_id, cluster_id) VALUES(?,?)" jarp -q "$QUERY" -a "$INSERT" -t 0.1 -c 0.3 -f 512Make sure that the
clusterstable exists and is empty before running the script. -
Clustering using the output of GenerateMD (in Unix)
generatemd c input.smi -c CF -k cfp.xml -D | jarp -f 512 -t 0.1 -c 0.3 -g -
Testing different
-cparameters. Using the output of NNeib. Singletons get negative cluster ids:generatemd c input.smi -k CF -c cfp.xml -D -o fingerprints.txt nneib -f 512 -t 0.1 -g <fingerprints.txt >neighborlists.txt -
Displaying the structures of the first cluster using the CreateView and MarvinView applications:
-
Clustering:
generatemd c input.sdf -k CF -c cfp.xml -D -o fingerprints.txt jarp -g -t 0.1 -c 0.3 -f 512 < fingerprints.txt > clusters.txt -
Creating an SDfile containing the structures from the first cluster (clid=1):
crview -i id -c "clid=1" -s input.sdf -t clusters.txt >jarp_result1.sdf -
Displaying the structures and the NSC field (it comes from the original SDfile):
mview --gridbag -c 3 -r 3 -f NSC jarp_result1.sdf
A screenshot of MarvinView showing the cluster:
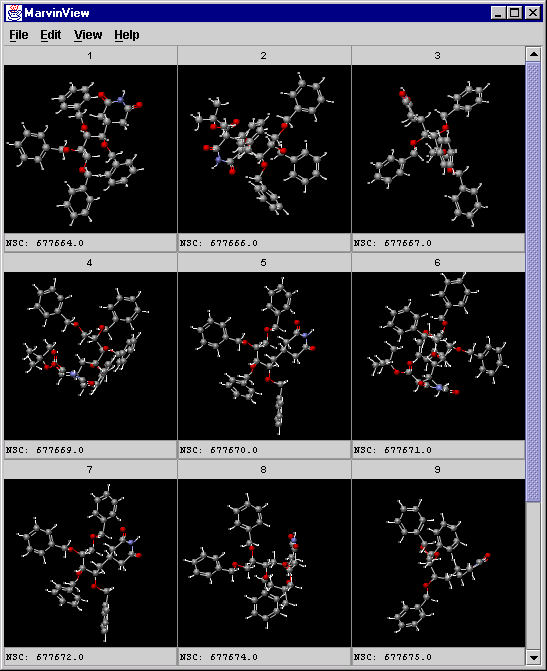
-
-
Displaying the central objects of clusters that contain at least 20 compounds (size >= 20) using the CreateView and MarvinView applications:
-
Clustering:
generatemd c input.sdf -k CF -c cfp.xml -D -o fingerprints.txt jarp -g -t 0.1 -c 0.3 -f 512 -x -z < fingerprints.txt > clusters.txt -
Creating an SDfile containing central objects of the clusters satisfying the condition:
crview -i "centr:2" -c "size>=20" -d "clid:size" -s input.sdf -t clusters.txt >jarp_result1.sdf -
Displaying the structures, the NSC field (comes from the original SDfile), and the cluster size (only for the central compounds):
mview -c 3 -r 3 -f "NSC:clid:size" jarp_result2.sdf
A screenshot of MarvinView showing the central objects:
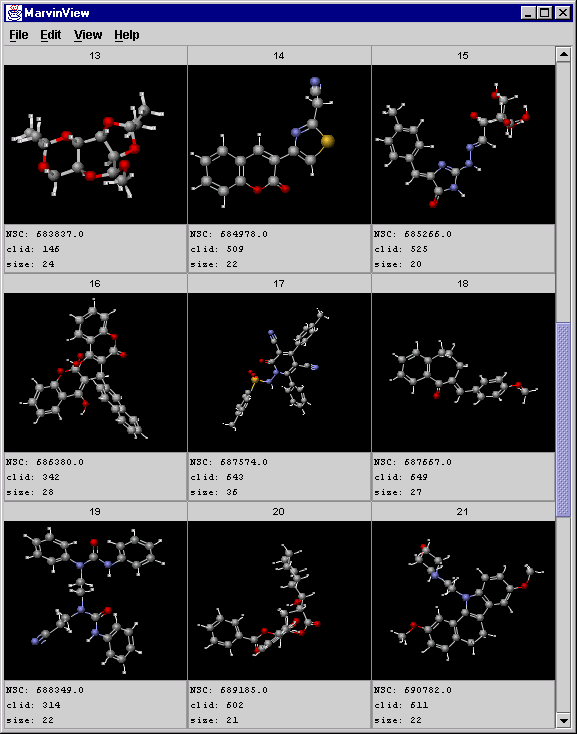
-
References
-
Jarvis, R. A.; Patrick, E. A. Clustering Using a Similarity Measure Based on Shared Nearest Neighbors IEEE Trans. Comput. 1973, C22, 1025-1034
-
Brown, R. D.; Martin. Y. C. Use of Structure-Activity Data To Compare Structure-Based Clustering Methods and Descriptors for Use in Compound Selection J. Chem. Inf. Comput. Sci. 1996, 36, 572-584
-
Barnard, J. M.; Downs, G. M. Chemical Fragment Generation and Clustering Software J. Chem. Inf. Comput. Sci. 1997, 37, 141-142